
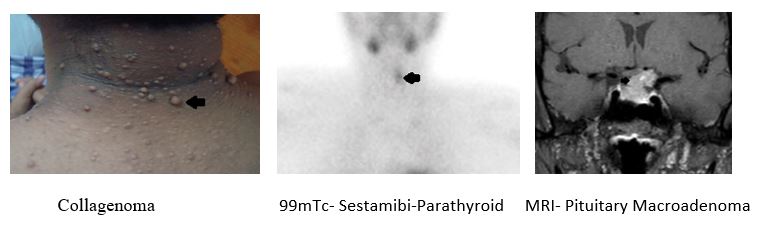
A 55-year-old lady complained of skin lesions on her anterior abdominal wall and neck, when she went to her Endocrinologist for a routine diabetic follow up. Examination revealed multiple firm hypopigmented papules, clinically suggestive of collagenomas, on the anterior abdominal wall. Retrospectively, she also had a history of bony pains, dyspepsia and early menopause. She had a significant family history with a prolactinoma in her daughter. She was then biochemically evaluated for MEN 1 syndrome as collagenomas are present in up to 70% of these patients. Evaluation for hyperparathyroidism showed serum calcium of 10.9mg/dl and PTH (parathyroid hormone) level of 155pg/mL with normal vitamin D and creatinine levels. DEXA scan showed significant osteopenia. Ultrasound neck and 99mTc- Sestamibi was suggestive of a doubtful enlargement of the left inferior parathyroid gland. Her prolactin level was found to be elevated at 270ng/mL.MRI pituitary showed a pituitary macroadenoma. Ultrasound abdomen did not reveal any obvious pancreatic or adrenal lesions. A diagnosis of MEN 1 was made with the presence of primary hyperparathyroidism, prolactinoma and collagenoma.
She underwent 3 and half parathyroidetomy with cervical thymectomy. Her intra operative PTH dropped from 150pg/mll to 25pg/ml, ten minutes post excision. Her serum calcium normalised and the post operative period was uneventful. The final biopsy was reported as parathyroid hyperplasia. She was started on cabergoline for management of the prolactinoma.
Two major forms of Multiple Endocrine Neoplasia, are recognized and are referred to as type 1 (MEN1) and type 2 (MEN2). Each form is characterized by the development of tumors within specific endocrine glands. MEN1 and MEN2 may be inherited as autosomal-dominant syndromes or they may occur sporadically, that is, without a family history.
The prevalence of MEN1 has been estimated from random post-mortem studies to be 0.25%, and about 1–18% in patients with primary hyperparathyroidism, 16 –38% in patients with gastrinomas, and less than 3% in patients with pituitary tumors. The disorder affects all age groups, with a reported age range of 5 to 81 yr. MEN1 is inherited as an autosomal dominant disorder with a high degree of penetrance such that clinical and biochemical manifestations of the disorder will have developed in 80% and greater than 98% of MEN1 patients, respectively, by the fifth decade.
Hypercalcemia resulting from multiple parathyroid tumors or parathyroid hyperplasia, is the presenting manifestation in 90% of patients with MEN1. Primary hyperparathyroidism reaches nearly 100% penetrance by age 50, with typical onset around age 20. Enteropancreatic tumors occur in 30-75% of patients with MEN1. The majority of the MEN1 enter pancreatic tumors are multifocal duodenal gastrinomas, often resulting in the Zollinger-Ellison syndrome of gastrin hypersecretion. Pituitary adenomas, most commonly prolactinomas are found in 15 to 20% of patients. Other tumors which occur with increased frequency in MEN 1 syndrome include carcinoids, adrenal tumors, angiomyolipomas, ependymomas, lipomas, meningiomas and cutaneous collagenomas and angiofibromas. The presence of collagenomas and angiofibromas is specific for MEN1 and suggest this diagnosis, when noticed in patients with hyperparathyroidism.
Dr Siddhartha Chakravarthy MS, MCh Consultant Endocrine & Breast Surgeon Apollo Hospital, Jubilee Hills Hyderabad
Dr Sreedevi Patnala MD, DM Consultant Endocrinologist Apollo Hospitals Secunderabad
