Osteosarkom oder osteogenes Sarkom ist die häufigste Knochenkrebsart. Osteosarkom ist eine Krebsart, bei der Tumorzellen unreifes Knochengewebe, sogenanntes Osteoid, produzieren. Obwohl Osteosarkom selten ist, ist es der häufigste Knochenkrebs bei Kindern. Osteosarkom befällt normalerweise die langen Knochen des Körpers in der Nähe der Wachstumsfugen, Bereiche, in denen sich bei jungen Menschen neues Gewebe bildet. Die meisten Tumoren treten rund um das Knie entweder im Oberschenkelknochen oder im Schienbein auf. Osteosarkom kann auch in anderen Körperteilen entstehen, sogar außerhalb der Knochen in den Weichteilen, insbesondere bei älteren Patienten. Mehr Männer als Frauen erkranken an diesem Krebs.



Übersicht
Was sind die häufigsten Arten von Knochenkrebs?
Es gibt verschiedene Arten von Knochentumoren. Sie werden nach dem Knochen- oder Gewebebereich benannt, in dem sie entstehen, und nach der Art der Zellen, die sie enthalten. Die am häufigsten vorkommenden Arten von primärem Knochenkrebs sind:
- Osteosarkom
-
- Chondrosarkom
-
Chondrosarkom ist Krebs der Knorpelzellen. Mehr als 40 % aller Knochenkrebserkrankungen bei Erwachsenen sind Chondrosarkome, was es zum häufigsten Knochenkrebs bei Erwachsenen macht. Das durchschnittliche Diagnosealter liegt bei 51 Jahren, und 70 % der Fälle betreffen Patienten über 40 Jahre. Chondrosarkom wird in der Regel in einem frühen Stadium diagnostiziert und ist oft von geringem Grad. Viele Chondrosarkomtumoren sind gutartig (kein Krebs). Tumoren können sich überall im Körper entwickeln, wo Knorpel vorhanden ist, insbesondere im Becken, Bein oder Arm.
- Ewing-Sarkom
-
Das Ewing-Sarkom ist die zweithäufigste Knochenkrebsart bei Kindern und Jugendlichen und die dritthäufigste bei Erwachsenen. Es macht etwa 8 % aller Knochenkrebserkrankungen bei Erwachsenen aus. Das Ewing-Sarkom kann in Knochen, Geweben oder Organen beginnen, insbesondere im Becken, der Brustwand, den Beinen oder Armen.
- Chordome
-
Ein Chordom ist eine seltene Art von Krebstumor, der überall entlang der zentralen Teile der Wirbelsäule auftreten kann, von der Schädelbasis bis zum Steißbein. Chordome sind langsam wachsende Tumoren, die aus Knochen- und Weichteilkomponenten bestehen. Sie treten nach der Behandlung häufig wieder auf und in etwa 20 bis 30 Prozent der Fälle breitet sich der Krebs auf andere Bereiche des Körpers aus, beispielsweise auf die Lunge oder die Knochen. Ungefähr die Hälfte aller Chordome tritt an der Basis der Wirbelsäule (Kreuzbein) auf, etwa ein Drittel an der Schädelbasis (Hinterkopf) und der Rest an den Hals-, Brust- oder Lendenwirbeln (unterer Rücken). Während das Chordom wächst, übt es Druck auf die angrenzenden Bereiche des Gehirns oder des Rückenmarks aus, was zu den Anzeichen und Symptomen führt. Ein Chordom an einer beliebigen Stelle entlang der Wirbelsäule kann Schmerzen, Schwäche oder Taubheitsgefühl im Rücken, den Armen oder Beinen verursachen. Chordome treten typischerweise bei Erwachsenen zwischen dem 40. und 70. Lebensjahr auf. Etwa 5 Prozent der Chordome werden bei Kindern diagnostiziert. Aus unklaren Gründen sind Männer etwa doppelt so häufig betroffen wie Frauen.
- Wie häufig sind Chordome?
-
Chordome sind selten und kommen jährlich bei etwa einem von einer Million Menschen vor. Chordome machen weniger als 1 Prozent aller Tumoren aus, die das Gehirn und das Rückenmark betreffen.
- Was sind die Ursachen eines Chordoms?
-
Veränderungen im TBXT-Gen werden mit Chordomen in Verbindung gebracht. Eine vererbte Duplikation des TBXT-Gens, die in einigen Familien festgestellt wurde, ist mit einem erhöhten Risiko für die Entwicklung eines Chordoms verbunden. Sowohl Duplikationen als auch eine erhöhte Expression des TBXT-Gens führen zur Produktion von überschüssigem Brachyury-Protein. Von den Tumorzellen exprimiertes Brachyury kann leicht durch Immunhistochemie identifiziert werden.
- Sekundärer (oder metastasierter) Knochenkrebs
-
Sekundärer (oder metastasierter) Knochenkrebs ist Krebs, der sich von einem anderen Körperteil auf den Knochen ausbreitet. Diese Art von Knochenkrebs kommt häufiger vor als primärer Knochenkrebs.
- Wie werden primäre Knochensarkome klassifiziert?
-
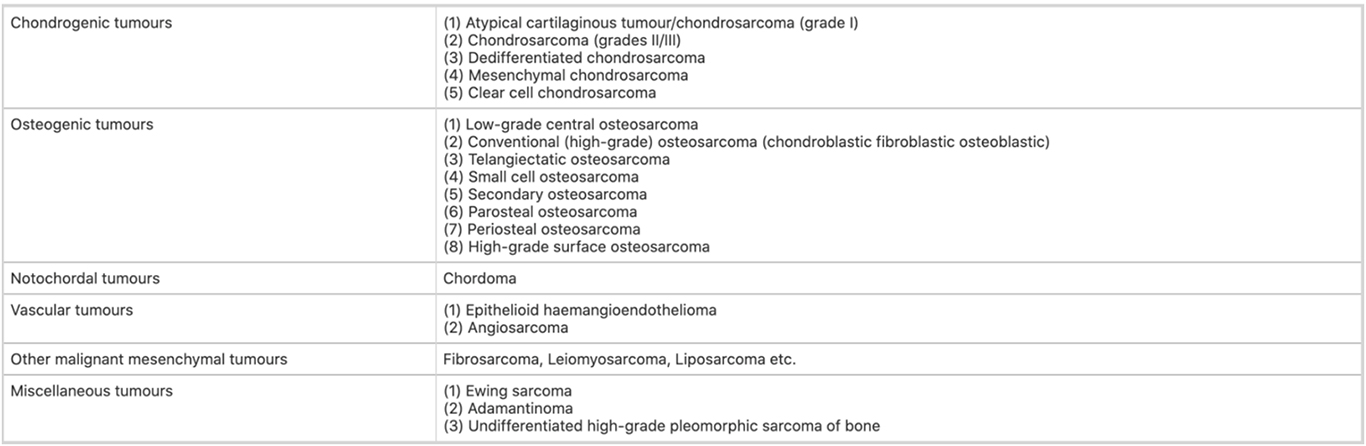
Nachfolgend finden Sie die WHO-Klassifikation primärer Knochensarkome.
- Wie werden Knochensarkome in Stadien eingeteilt?
-
Alle Knochensarkome werden gemäß dem folgenden AJCC/TNM-Staging-System eingeteilt.

Symptome
Bei Patienten mit einem Knochentumor treten häufig eines oder mehrere der folgenden Symptome auf:
- Schmerzen im Bereich des Tumors werden im Allgemeinen als dumpf und stechend beschrieben. Sie können sich nachts verschlimmern und bei Aktivität zunehmen.
- Fieber und Nachtschweiß
- Viele Patienten haben keinerlei Symptome, bemerken stattdessen eine schmerzlose Masse.
- Obwohl Knochentumore nicht durch ein Trauma verursacht werden, kann eine Verletzung manchmal dazu führen, dass ein Tumor Schmerzen verursacht. Eine Verletzung kann auch dazu führen, dass ein durch einen Tumor geschwächter Knochen bricht. Dies kann sehr schmerzhaft sein.
- Gelegentlich können Tumoren zufällig entdeckt werden, wenn aus einem anderen Grund, beispielsweise aufgrund einer Knöchel- oder Knieverletzung, eine Röntgenaufnahme gemacht wird.
Risikofaktoren
Bei den meisten Patienten mit Knochentumoren sind keine Risikofaktoren bekannt. Es gibt jedoch eine Reihe von Risikofaktoren für Knochenkrebs, darunter auch genetische Faktoren. Menschen mit chronischen entzündlichen Erkrankungen wie Morbus Paget sind einem erhöhten Risiko ausgesetzt.
Weitere Risikofaktoren für die Entwicklung von Knochenkrebs sind:
- Unter 20 Jahre alt sein
- Strahlenbelastung, z. B. bei einer Strahlentherapie gegen eine andere Krebserkrankung
- Ein früherer Empfänger einer Knochenmarktransplantation
- Einen nahen Verwandten mit Knochenkrebs haben
- Personen mit erblichem Retinoblastom, einer Art von Augenkrebs, der am häufigsten bei Kindern auftritt.
Diagnose
In spezialisierten Zentren wie dem APCC werden die Patienten einer gründlichen Untersuchung durch den Arzt unterzogen. Anschließend werden eine oder mehrere der zur Diagnose von Knochentumoren verwendeten Instrumente eingesetzt:
- Röntgenaufnahmen sind oft die ersten Untersuchungen. Manchmal werden spezielle Aufnahmen angeordnet, um Knochentumoren besser darstellen zu können.
- Eine Biopsie kann entweder eine Nadelbiopsie oder eine chirurgische Biopsie sein. Viele Überlegungen müssen berücksichtigt werden, bevor eine Biopsie von einem Knochentumor durchgeführt wird, insbesondere von Krebstumoren. Ein multidisziplinäres Tumorgremium bespricht den besten Ansatz, um bei Bedarf eine Biopsie am APCC durchzuführen. Eine schlecht geplante oder durchgeführte Knochenbiopsie kann weitere Behandlungen gefährden.
- CT-Scans werden normalerweise verwendet, um eine erste Knochenkrebsdiagnose zu stellen und festzustellen, ob sich der Krebs auf andere Bereiche des Körpers ausgebreitet hat. CT-Scans können auch verwendet werden, um die Biopsienadel zu führen.
- Knochenszintigraphie – Eine Knochenszintigraphie mit Radionukliden kann zur Diagnose und Einstufung von Knochenkrebs verwendet werden. Dieses Mittel zur Erkennung von Knochenkrebs kann zeigen, ob sich der Primärtumor auf andere Stellen im Knochen ausgebreitet hat und wie viel Schaden er verursacht hat. Bei einer Knochenszintigraphie wird eine kleine Dosis radioaktiven Materials in ein Blutgefäß injiziert, wo es durch den Blutkreislauf wandert. Das Material sammelt sich dann in den Knochen und wird von einem Scanner durch nukleare Bildgebung erkannt. Dieser Test ist sehr empfindlich und kann kleine Metastasen finden, bevor sie auf einer normalen Röntgenaufnahme erscheinen würden. Andere Erkrankungen wie Arthritis oder Infektionen sehen auf der Szintigraphie jedoch ähnlich aus, sodass häufig eine bestätigende Biopsie erforderlich ist.
- Mithilfe einer MRT kann ein Tumor im Knochen abgegrenzt werden und es kann auch festgestellt werden, ob sich Krebszellen ins Gehirn oder Rückenmark ausgebreitet haben. Außerdem wird sie zur Beurteilung der Behandlungsreaktion und bei der Nachsorge eingesetzt. Bei Wirbelsäulentumoren ist die MRT wahrscheinlich die beste Untersuchungsmethode.
- Die PET-CT-Untersuchung ist wahrscheinlich das zuverlässigste Verfahren zur Krebserkennung. Sie ist ein wichtiges Instrument zur Stadienbestimmung der Krankheit vor der Behandlung sowie zur Überwachung nach der Behandlung.
- Darüber hinaus werden auch andere Tests wie Blutuntersuchung, alkalische Phosphatase, BSG, Knochenmarkzytologie oder -biopsie usw. durchgeführt.
- Die Pathologie einschließlich der Immunhistochemie ist für jeden Patienten mit Knochensarkomen von entscheidender Bedeutung.
- Genetische und molekulare Untersuchungen wie die EWS-FLI1-Translokation bei Ewing-Sarkomen und anderen Sarkomen sind für eine Diagnose äußerst wichtig.
Behandlung
- MDT für Knochentumoren
-
Zusätzlich zur Versorgung oder Überwachung durch ein spezialisiertes MDT für Knochensarkom sollte den Patienten ein Betreuer zugewiesen werden. Kinder, Teenager und junge Erwachsene sollten auch im entsprechenden MDT für Kinder oder TYA (junge Erwachsene) besprochen werden. Dies erfordert ausreichend Fachpersonal, um eine altersgerechte Versorgung zu gewährleisten. Ein MDT für Knochensarkom sollte ordnungsgemäß aufgebaut sein, die Anforderungen für die Kernmitgliedschaft der entsprechenden Fachrichtungen einhalten und Mindestkriterien für die Anzahl der jährlich behandelten Patienten erfüllen; es sollte wie vereinbart Daten zu Patienten, Tumoren, Behandlung und Ergebnissen erfassen.
- Chirurgie bei Knochensarkomen und Extremitätenerhaltungschirurgie
-
Entscheidungen über das optimale chirurgische Verfahren für den Primärtumor (d. h. Gliedmaßenerhalt oder Amputation) erfordern eine MDT-Diskussion unter Berücksichtigung der Tumorgröße und der Beteiligung anatomischer Strukturen, der Reaktion auf neoadjuvante Therapien und der Patientenpräferenz. Die chirurgische Rekonstruktion kann von der Wahl des Patienten und des Chirurgen beeinflusst werden und sollte einer offenen Diskussion der Risiken und Vorteile der verfügbaren Optionen und der erwarteten funktionellen Ergebnisse folgen.
Die Operation bei Knochensarkomen erfolgt in 3 Schritten.
- Resektion des Tumors mit ausreichenden Rändern.
- Rekonstruktion des Knochendefekts, der durch die Entfernung des Tumors entstanden ist.
- Rekonstruktion des Weichgewebes und Weichteilausgleich, um eine frühzeitige Wiederherstellung der Funktionsfähigkeit zu ermöglichen.
Ziel der kurativen Chirurgie ist die Entfernung des gesamten Tumors mit ausreichenden Rändern. Wenn möglich, sollte eine breite En-bloc-Resektion des betroffenen Knochenteils und des betroffenen Weichteilgewebes durchgeführt werden. Enge chirurgische Ränder können mit (MRT-inerten) Clips markiert werden, die im Operationsfeld platziert werden. Bei Knochensarkomen sollte die Operation, soweit möglich, die Entfernung aller anatomischen Strukturen umfassen, die zum ursprünglichen Tumorvolumen vor der Chemotherapie gehörten. Die Probe sollte so ausgerichtet sein, dass der Pathologe die anatomische Lage und Dicke der chirurgischen Ränder beschreiben kann.
Die Knochenrekonstruktion nach der Entfernung des Tumors kann je nach Lage des Tumors und Alter des Patienten biologisch oder nicht-biologisch erfolgen. Bei nicht-biologischen Rekonstruktionen wird eine Tumormegaprothese verwendet, die eine sofortige Wiederherstellung der Funktion nach der Operation ermöglicht. Bei der biologischen Rekonstruktion werden Allograft-Knochen oder Autograft-Knochen verwendet, wobei der Knochen entfernt, durch eine einzelne hochdosierte Strahlendosis von Tumorzellen befreit und nach Stabilisierung mit geeigneten Implantaten wieder an seinen Platz zurückverpflanzt werden kann.
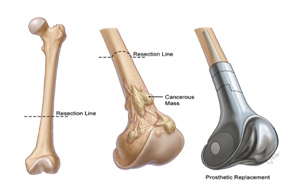
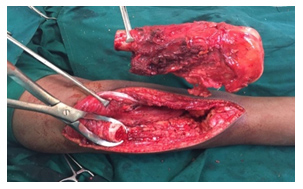
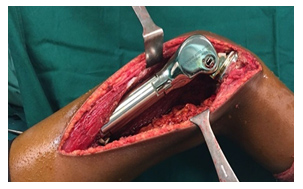
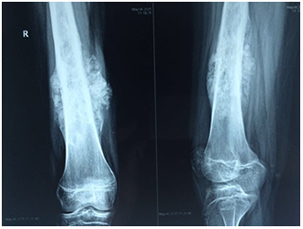
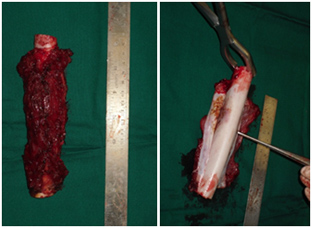
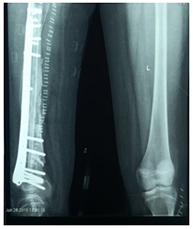
- Systemische Therapie (Chemotherapie/zielgerichtete Therapie)
-
Osteosarkom
Osteogenes Sarkom ist einer der häufigsten Knochentumoren und birgt ein hohes Risiko, sich in die Lunge auszubreiten. Daher ist neben einer lokalen Therapie wie Resektion auch eine Chemotherapie erforderlich, um Fernmetastasen zu verhindern. Sie gelten als chemoresistente Tumoren und normalerweise wird eine Kombinationschemotherapie angewendet. Es werden hauptsächlich zwei Behandlungsschemata angewendet: Hochdosiertes Methotrexat und Kombinationstherapie mit Ifosfamid/Cisplatin/Doxorubicin. Die Chemotherapiezyklen werden normalerweise vor der Operation als neoadjuvante Chemotherapie und nach der Operation als adjuvante Chemotherapie verabreicht. Beide Behandlungsschemata erfordern unterstützende Medikamente wie Filgrastim-Unterstützung, Antiemetika usw., um die Wahrscheinlichkeit von Nebenwirkungen zu verringern. Im Fall von metastasiertem Osteosarkom umfasst die Erstbehandlung Chemotherapie und Neubewertung. Wenn bei der Neubewertung die metastasierten Läsionen (Lunge) resektabel sind, werden sie entfernt und die Chemotherapie wird fortgesetzt.
Ewing-Sarkom
Chemotherapie ist Teil der Standardbehandlung für Ewing-Sarkome. Sie kommen normalerweise bei Jugendlichen und jungen Erwachsenen vor. Nach vollständiger Stadienbestimmung wird eine Induktionschemotherapie durchgeführt und anschließend eine lokale Behandlung durchgeführt. Die lokale Behandlung erfolgt entweder durch Operation (bei peripheren Läsionen) oder Strahlentherapie (Beckenläsionen) oder beides (bei Brustwandtumoren). Die gesamte Behandlungsdauer ist lang: 9-12 Monate gemäß den Behandlungsprotokollen. Normalerweise wird die Behandlung von jungen Patienten gut vertragen. Nach der Behandlung ist eine langfristige Nachsorge sehr wichtig, damit etwaige langfristige behandlungsbedingte Nebenwirkungen umgehend behandelt werden können. Bei richtiger Behandlung handelt es sich um Tumore mit hoher Heilungsrate.
Andere Knochentumoren
Die Behandlung von Chondrosarkomen und den meisten anderen gutartigen Knochentumoren erfolgt überwiegend chirurgisch, obwohl bei dedifferenzierten und mesenchymalen Subtypen auch Chemotherapie eine Rolle spielen kann. Sie gelten in der Regel als chemoresistent. Bei niedriggradigen Chondrosarkomen ist auch eine Beobachtung möglich, sofern keine Symptome auftreten.
Derzeit spielt die zielgerichtete Therapie bei Knochentumoren keine große Rolle
- Strahlentherapie bei Knochensarkomen
-
Strahlentherapie wird häufig zur definitiven Behandlung des Primärtumors beim Ewing-Sarkom eingesetzt, aber die relative Strahlenresistenz von Osteosarkom und Chondrosarkom bedeutet, dass sie nur dann als definitive Behandlung eingesetzt wird, wenn keine chirurgische Option besteht. Diese Tumoren erfordern typischerweise sehr hohe Strahlendosen (>66 Gy) für eine dauerhafte Kontrolle. Die Angrenzung an empfindliche Strukturen, insbesondere an der Schädelbasis, der Wirbelsäule und dem Becken, erfordert eine hochpräzise Strahlentherapie, insbesondere wenn eine vollständige Resektion nicht möglich scheint.
Eine Strahlentherapie wird postoperativ nicht routinemäßig durchgeführt, kann jedoch in bestimmten Fällen mit hohem Risiko oder in Situationen wie Chordomen eingesetzt werden. Bei allen Tumorarten spielt die Strahlentherapie auch eine palliative Rolle.
- Protonentherapie bei Knochensarkomen
-
Protonen oder Kohlenstoffionen, oft in Kombination mit Photonen, werden zunehmend zur Behandlung nicht resektabler primärer Knochensarkome eingesetzt.
Hervorragende Ergebnisse werden bei Chondrosarkomen und Chordomen der Schädelbasis berichtet, bei denen eine Protonenbestrahlung in Kombination mit einer Operation lokale Kontrollraten von etwa 70–90 % erreichen kann. Bei nicht oder unvollständig resektablen Osteosarkomen betrug das krankheitsfreie 65-Jahres-Überleben (DFS) 5 % und das 67-Jahres-Gesamtüberleben (OS) XNUMX %. Hohe lokale Kontrollraten wurden auch bei sakralen Chordomen erreicht.
- Vorteile der Protonentherapie bei Knochensarkomen
-
- Höhere Kontrollraten mit Protonentherapie im Vergleich zu IMRT/SRS/Cyberknife. Eine Studie der National Cancer Database aus den Vereinigten Staaten hat gezeigt, dass die Kontrollraten bei Patienten, die mit Protonentherapie behandelt werden, im Vergleich zu herkömmlichen Photonentechniken wie IMRT, IGRT, SRS/Cyberknife usw. dramatisch besser sind.
- Niedrige Raten akuter und später Toxizitäten. Da höhere Dosen verabreicht werden müssen, sind herkömmliche Bestrahlungstechniken mit einer relativ größeren Belastung durch Nebenwirkungen verbunden. In bestimmten Situationen wird die Strahlendosis sogar reduziert, um die Wahrscheinlichkeit von Nebenwirkungen zu begrenzen. Die Protonentherapie gewährleistet jedoch die sichere Verabreichung höherer Strahlendosen bei minimaler Toxizität.
- Reduziertes Risiko einer Zweitkrebserkrankung. Strahlentherapie in Kombination mit Chemotherapie ist häufig mit einem erhöhten Risiko für Sekundärkrebs verbunden. Mehrere Studien haben gezeigt, dass die Protonentherapie die Wahrscheinlichkeit für Sekundärkrebs deutlich verringert. Diese Eigenschaft ist insbesondere bei jüngeren Patienten von großem Nutzen.
- Vergleich mit IMRT/SRS/Cyber Knife
-
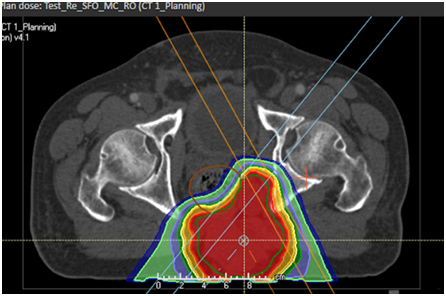
VMAT-Plan für ein sakrales Chordom
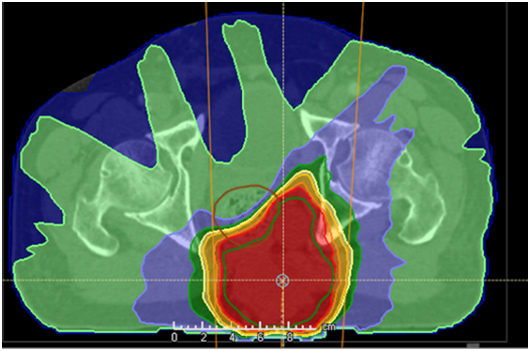
- Protonentherapieplan (IMPT) für ein sakrales Chordom
-
Demonstriert die Schonung von Harnblase, Rektum, Beckenknochen und Knochenmark.

Andere Knochen- und Weichteiltumoren

Copyright © 2026 Apollo Proton Cancer Centre. Alle Rechte vorbehalten